

DeepMind AlphaFold protein database alphabet provides a revolutionary resource for understanding protein structures. This database, built on the groundbreaking AlphaFold methodology, offers unparalleled detail and accuracy in predicting protein structures. It’s a game-changer for biological research, pushing the boundaries of our understanding of life’s building blocks.
This comprehensive database delves into the intricacies of protein structure prediction, offering a fascinating look at the methodology behind AlphaFold. From its historical context to its impact on various scientific disciplines, this resource unveils a wealth of information. The database’s structure, data content, and features are explored in detail, revealing how researchers utilize it to study protein function, facilitate drug discovery, and drive advancements in biotechnology.
Challenges, limitations, and future directions are also addressed, presenting a holistic view of this powerful tool.
Introduction to DeepMind AlphaFold Protein Database
The DeepMind AlphaFold protein database represents a monumental leap forward in our ability to understand and model proteins. This database, built on the AlphaFold algorithm, provides predicted 3D structures for a vast collection of proteins, dramatically accelerating biological research and potentially revolutionizing drug discovery and biotechnology. The accuracy and speed of prediction are truly remarkable, enabling scientists to study protein function and interactions with unprecedented detail.AlphaFold leverages a powerful machine learning approach to predict protein structures, vastly improving upon traditional methods that were often slow and computationally intensive.
This innovative technology has the potential to address significant challenges in biology and medicine. The database is freely accessible, fostering collaboration and accelerating scientific discovery across diverse fields.
Methodology Behind Protein Structure Prediction in AlphaFold
AlphaFold’s groundbreaking approach to protein structure prediction rests on a sophisticated machine learning model. It employs a deep learning neural network trained on vast amounts of protein sequence and structure data. This training allows the network to learn the intricate relationships between amino acid sequences and the resulting 3D structures. Crucially, AlphaFold integrates evolutionary information into its predictions, taking into account the evolutionary history of protein families.
This approach is far more efficient and accurate than previous methods, which often relied on experimental data or computational heuristics.
Significance of the Database for Biological Research
The AlphaFold database is a treasure trove of information for biological researchers. It allows scientists to visualize the 3D structures of proteins, understanding their shapes and how they interact with other molecules. This knowledge is crucial for comprehending protein function, identifying potential drug targets, and designing new therapies. Researchers can investigate protein-protein interactions, enzyme mechanisms, and the role of proteins in various biological processes.
The availability of predicted structures significantly accelerates research by enabling researchers to focus on functional analysis and validation rather than time-consuming structure determination experiments.
Historical Context of Protein Structure Prediction
Historically, determining protein structures was a painstaking and time-consuming process. Traditional methods, like X-ray crystallography and nuclear magnetic resonance (NMR), were limited by experimental constraints. They required specialized equipment, were often expensive, and were applicable to only a fraction of the proteins known. The lack of complete structural information significantly hampered our understanding of biological processes. The development of AlphaFold marks a paradigm shift, transitioning from an experimental bottleneck to a powerful predictive tool.
Impact of AlphaFold on Various Scientific Disciplines
AlphaFold’s impact extends across numerous scientific disciplines. In drug discovery, it accelerates the identification of potential drug targets and the design of novel therapeutic molecules. In materials science, it assists in the development of new bio-inspired materials. In agriculture, it aids in understanding crop protein function and enhancing crop yield. In structural biology, it empowers researchers to investigate protein interactions and mechanisms at a level of detail previously unimaginable.
For example, the accurate prediction of protein structures for a particular enzyme involved in a metabolic pathway can reveal insights into its catalytic activity, leading to the development of novel strategies for controlling or enhancing the process. The impact is evident in various applications, from disease modeling to biotechnology.
Data Structure and Format: Deepmind Alphafold Protein Database Alphabet
The AlphaFold Protein Database provides a comprehensive resource for researchers studying protein structures. Its well-defined structure and format enable efficient access and analysis of predicted protein structures, facilitating a deeper understanding of biological processes. This organization allows for effective querying and retrieval of specific protein information, contributing significantly to biological research.The database’s structure is meticulously designed to allow researchers to locate and retrieve specific protein information easily.
DeepMind’s AlphaFold protein database alphabet is fascinating, mapping amino acid sequences to 3D structures. It’s like a secret code, revealing the intricate forms of proteins. This complexity reminds me of how a particular fragrance can evoke the uneasy chill of encountering an apparition, as explored in this insightful article: the fragrance evokes the uneasy chill of encountering an apparition.
Understanding these intricate protein structures, much like deciphering the fragrance’s hidden message, opens a whole new world of biological possibilities.
This facilitates both broad exploration of protein structures and focused investigation of particular protein families or characteristics. This organized structure is critical for the effective use of the database by the scientific community.
Database Table Structure
The AlphaFold database is not presented as a single, monolithic table. Instead, the data is organized across multiple tables, each dedicated to a specific aspect of the protein information. This modular approach allows for flexibility and scalability, accommodating the diverse types of data associated with protein structures. This structure enables efficient storage and retrieval of information related to various aspects of proteins.
| Table Name | Description |
|---|---|
| Protein Sequence | Stores the amino acid sequences of proteins. |
| Predicted Structure | Contains the predicted 3D structure of each protein. |
| Protein Metadata | Provides supplementary information about the protein, such as its source organism, function, and associated diseases. |
Data Format for Protein Structures
The predicted protein structures are typically represented in a standardized format that allows for efficient processing and visualization. This standard format enables interoperability between different software tools and platforms, facilitating analysis and comparison of protein structures.
| Format | Description |
|---|---|
| PDB (Protein Data Bank) format | A widely used format for storing 3D protein structures. It specifies the coordinates of atoms in the protein, along with other relevant information like secondary structure elements. |
| AlphaFold-specific JSON format | A format tailored to the AlphaFold database, enabling efficient storage and retrieval of the detailed structure predictions, including various aspects of the predicted structures. |
File Types in the Database
The AlphaFold database utilizes various file types to accommodate the different kinds of data associated with protein structures. This approach ensures that the database is versatile and adaptable to the needs of researchers.
- JSON files: These files store metadata, sequences, and predicted structures in a structured format, enabling easy parsing and manipulation by computer programs.
- PDB files: These files are frequently used to represent 3D structures in a standardized format. This allows researchers to easily import the structures into various visualization and analysis software.
- FASTA files: These files are used to store protein sequences. They provide a simple text-based format for representing the amino acid sequence of a protein, facilitating quick access to the sequence data.
Unique Protein Identification
Unique identification of proteins is crucial for maintaining data integrity and enabling accurate referencing across different datasets.
- Protein ID: A unique identifier assigned to each protein entry in the database. This ID serves as a key for locating specific protein information within the database.
- Sequence ID: A unique identifier based on the protein’s amino acid sequence, ensuring that proteins with identical sequences are correctly identified.
Standardized Formats like PDB
The use of standardized formats like PDB is essential for interoperability and data exchange. It allows researchers to share and analyze protein structures using a common language, promoting collaboration and the advancement of scientific knowledge.
PDB (Protein Data Bank) format ensures consistent representation and interpretation of protein structures across different research groups and software applications.
Data Content and Features
The AlphaFold Protein Database is a treasure trove of protein structure information, meticulously curated and predicted using DeepMind’s AlphaFold technology. This detailed data allows researchers to visualize and analyze protein structures, furthering our understanding of biological processes and enabling advancements in medicine and materials science. Beyond just the structures, the database provides valuable metadata, further enriching the understanding of each protein.The database is not just a collection of 3D models; it’s a comprehensive resource for studying protein structure, function, and evolution.
Each entry provides a wealth of information, facilitating deeper investigation into the intricacies of life at the molecular level. This allows researchers to make informed predictions and hypotheses about the relationships between protein structure and function, leading to significant breakthroughs in various scientific fields.
Types of Information Stored
The database stores a diverse range of information for each protein structure, including the amino acid sequence, predicted 3D structure (atomic coordinates), and associated metadata. This rich dataset allows researchers to analyze proteins in unprecedented detail, correlating structure with function and evolutionary relationships. This detailed information is critical for various research applications, including drug discovery, materials science, and basic biological research.
Comparison of Different Protein Structures, Deepmind alphafold protein database alphabet
A crucial aspect of the database is the ability to compare different protein structures. This comparison facilitates the identification of similarities and differences, providing insights into evolutionary relationships and functional variations. Understanding these relationships is vital in identifying conserved structural motifs, which can provide clues to the protein’s function.
| Protein Type | Structure Features | Example |
|---|---|---|
| Enzyme | Highly specific active sites, crucial for catalytic reactions | Lysozyme, a bacterial enzyme |
| Structural Protein | Provides mechanical support, maintaining cell shape and integrity | Collagen, a major component of connective tissues |
| Transport Protein | Facilitates the movement of molecules across membranes | Hemoglobin, carrying oxygen in the blood |
Methods for Verifying Accuracy
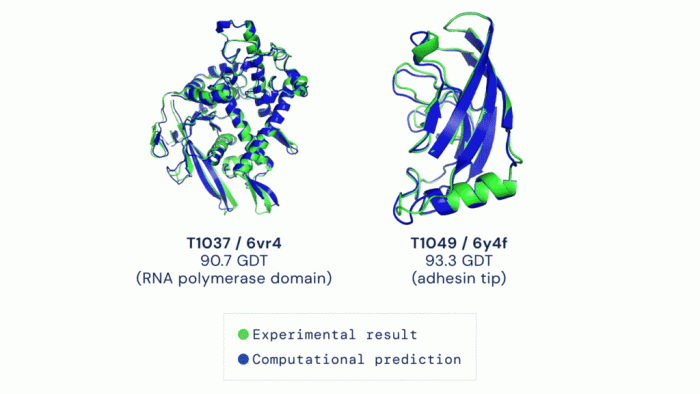
The accuracy of the predicted protein structures is meticulously verified using various methods. These methods include comparing the predicted structures to experimentally determined structures (crystallography or NMR) and evaluating the overall quality of the prediction using metrics such as the root-mean-square deviation (RMSD). These validation techniques are essential to ensure the reliability of the data.
“The use of multiple validation methods enhances the confidence in the predicted structures, ensuring the reliability and accuracy of the database.”
Examples of Proteins Present
The database contains a wide range of proteins, encompassing diverse functions and evolutionary lineages. This comprehensive representation is crucial for understanding the complexity of biological systems. Examples include proteins involved in metabolism, signaling, and immunity. Further, proteins related to specific diseases, such as those associated with cancer, are also represented in the database.
Accessing and Exploring the Database
The AlphaFold Protein Database is accessible through a variety of platforms and tools. These resources allow researchers to search for specific proteins, visualize their structures, and download the data. The user-friendly interfaces make it straightforward to navigate the extensive dataset and extract the desired information.
Applications and Uses of the Database
The DeepMind AlphaFold Protein Database provides a revolutionary resource for researchers, enabling unprecedented insights into protein structures and their functions. This detailed structural information fuels advancements across diverse scientific disciplines, from drug discovery to biotechnology. The database’s accessibility and comprehensiveness empower scientists to unravel complex biological mechanisms and accelerate the development of innovative solutions.The database’s profound impact lies in its ability to translate the language of protein structure into actionable knowledge.
This structural information is the key to understanding how proteins work, interact, and ultimately influence the intricate processes within living organisms. Scientists can now leverage this wealth of data to study protein function in unprecedented detail.
Protein Function Studies
The AlphaFold database facilitates in-depth investigations into protein function. Researchers can utilize the predicted 3D structures to understand how amino acid sequences translate into specific protein conformations and activities. This knowledge is crucial for understanding the roles proteins play in various cellular processes, including enzymatic catalysis, signaling pathways, and protein-protein interactions. For example, by examining the 3D structure of an enzyme, researchers can identify the active site and understand the mechanism of substrate binding and catalysis.
This knowledge is pivotal for designing new inhibitors or activators of specific enzymatic pathways.
Drug Discovery and Development
The database plays a pivotal role in accelerating drug discovery and development. Scientists can use the predicted 3D structures to identify potential drug targets and design small molecule inhibitors or activators. By understanding the precise interactions between a drug candidate and its target protein, researchers can optimize drug efficacy and minimize side effects. For instance, the 3D structure of a protein implicated in a disease pathway can help design drugs that specifically bind to and inhibit that protein, potentially alleviating disease symptoms.
This process, known as structure-based drug design, is greatly enhanced by the availability of the AlphaFold database.
Advancements in Biotechnology
The database’s impact extends to biotechnology, enabling the design and engineering of novel proteins with tailored functions. Researchers can use the predicted structures to design proteins with improved stability, activity, or specificity for specific applications. This is particularly useful in fields like biocatalysis, where enzymes with enhanced catalytic properties can be engineered for industrial processes. For example, designing an enzyme with a modified active site can allow for enhanced substrate specificity or reaction rates, thereby increasing efficiency in biofuel production or other biotechnological applications.
DeepMind’s AlphaFold protein database alphabet is fascinating, detailing the intricate structures of proteins. While I’m digging into the complexities of amino acid sequences, I’m also keeping an eye on the Twitter edit button blue test, which is a pretty cool development in social media. Ultimately, the precision of the AlphaFold database is a testament to the power of computational biology, and I’m excited to see how it will continue to impact our understanding of biological processes.
twitter edit button blue test It seems like a small change, but it has the potential to revolutionize the way we communicate on the platform. DeepMind’s work continues to inspire me.
Understanding Disease Mechanisms
The database enables researchers to gain a deeper understanding of disease mechanisms. By examining the 3D structures of proteins involved in disease pathways, researchers can identify key interactions and potential vulnerabilities. This knowledge is crucial for developing novel diagnostic tools, therapies, and preventative strategies. For example, studying the 3D structure of a protein mutated in a cancer patient can highlight specific regions of the protein that are altered and contribute to uncontrolled cell growth, paving the way for targeted therapies.
Workflow in Structural Genomics
The following workflow demonstrates how the database is used in structural genomics, a field focused on determining the 3D structures of large numbers of proteins.
| Step | Description |
|---|---|
| 1. Protein Selection | Researchers select a set of proteins based on their biological importance or potential role in a specific disease pathway. |
| 2. AlphaFold Prediction | The selected protein sequences are submitted to the AlphaFold database for prediction of their 3D structures. |
| 3. Structural Analysis | The predicted 3D structures are analyzed to identify key structural features, active sites, and potential interaction partners. |
| 4. Validation and Refinement | Experimental methods (e.g., X-ray crystallography or NMR) are used to validate and refine the predicted structures, thereby increasing accuracy and confidence in the findings. |
| 5. Functional Characterization | The validated structures are used to study protein function, design new drugs, or investigate disease mechanisms. |
Challenges and Limitations
The DeepMind AlphaFold protein database, while a monumental achievement, is not without its limitations. Maintaining such a vast and constantly evolving dataset presents unique challenges. Furthermore, the accuracy and reliability of protein structure predictions are not absolute, and the computational demands for analysis can be significant. Understanding these limitations is crucial for interpreting and applying the data effectively.
Maintaining and Updating the Database
The sheer volume of protein structures in the AlphaFold database, and the continuous influx of new data, necessitate robust systems for maintenance and updating. Data integrity, version control, and ensuring data consistency across different updates pose significant challenges. Efficient indexing and search mechanisms are also vital to allow researchers to quickly locate and retrieve relevant structures. The constant need to incorporate new experimental data and refine predicted structures adds another layer of complexity to the maintenance process.
DeepMind’s AlphaFold protein database alphabet is fascinating, detailing the intricate structures of proteins. While that’s incredibly useful for scientific research, it’s also important to consider the ethical implications of AI, like those raised by the recent controversy surrounding potential deepfakes of Congress members and lawmakers, explored in detail at microsoft deepfake congress lawmakers ai fraud. Ultimately, understanding these advancements in protein folding, and the potential for misuse in areas like AI-generated media, is crucial for responsible development and deployment of powerful technologies like AlphaFold.
Limitations of Protein Structure Prediction Methods
AlphaFold, despite its remarkable success, is not a perfect predictor of protein structures. Certain protein families, particularly those with intrinsically disordered regions or complex conformational changes, may be more challenging to model accurately. Furthermore, the accuracy of prediction often depends on the quality and quantity of training data available for specific protein sequences. In some cases, the predicted structures may not perfectly reflect the native conformation of the protein in its natural environment.
For instance, some structures may not account for post-translational modifications that can significantly alter protein function.
Accuracy and Reliability of Predicted Structures
The accuracy of AlphaFold predictions varies depending on the specific protein sequence and the available training data. While the overall accuracy is impressive, there are cases where the predicted structures may deviate from experimentally determined structures. Researchers should critically evaluate the reliability of predicted structures, particularly when making inferences about protein function or developing new therapies. Confidence scores are often associated with each prediction, indicating the level of certainty in the model’s output.
Scientists should use these scores as a guide for interpreting the results.
Computational Resources for Database Usage
Analyzing and utilizing the AlphaFold database requires substantial computational resources. The sheer size of the database necessitates powerful servers and high-bandwidth networks. Researchers might need to use specialized software and tools optimized for handling large datasets and complex analyses. Moreover, computational resources are essential for running simulations and performing complex calculations on the predicted structures. This demand for computational power can present a barrier for some researchers.
Ongoing Research Efforts in Improving AlphaFold
Ongoing research efforts are focused on improving the accuracy and efficiency of AlphaFold predictions. These efforts include:
- Developing more sophisticated machine learning models to better capture the complex relationships between protein sequences and structures.
- Improving the training data used to train AlphaFold, including incorporating more diverse protein families and experimental data.
- Incorporating new biological insights and constraints into the prediction process, such as evolutionary information and knowledge of protein interactions.
- Developing more efficient algorithms for processing and analyzing the vast amount of data in the AlphaFold database.
These advancements will further enhance the utility and reliability of the AlphaFold database in the future.
Future Directions and Potential

The AlphaFold protein database, a revolutionary resource, is poised for significant expansion and evolution. Its current utility in biological research is undeniable, but the potential for its application extends far beyond the realm of biology. This exploration delves into the exciting future directions, highlighting potential uses in diverse fields and emphasizing the importance of integration with other biological datasets.
Expanding the Database’s Scope
The current AlphaFold database primarily focuses on protein structures. Future iterations could incorporate additional data, such as protein-protein interaction information, post-translational modifications, and cellular localization data. This expanded scope would create a more comprehensive view of protein function, providing a holistic understanding of protein behavior within the intricate cellular environment.
Integration with Other Biological Resources
Integrating AlphaFold with existing databases, like Gene Ontology and UniProt, will allow for richer analysis. This combined data will facilitate the identification of functional relationships between proteins and the elucidation of complex biological pathways. For example, researchers could directly link a predicted protein structure from AlphaFold to its known function in Gene Ontology, accelerating the discovery process. This interconnectedness fosters a more comprehensive and integrated understanding of biological systems.
Enhanced Protein Structure Prediction Methodologies
Emerging methodologies in protein structure prediction hold the potential to further refine the accuracy and efficiency of AlphaFold. Machine learning techniques are constantly evolving, and advancements in deep learning algorithms may lead to even more accurate predictions, especially for complex protein structures or those lacking sufficient experimental data. Deep learning models trained on diverse datasets could potentially improve the accuracy of predictions for protein structures in previously challenging cases.
Potential Applications Beyond Biology
The applications of AlphaFold extend beyond biology to various fields. The database’s ability to predict protein structures can be leveraged in materials science, where novel protein-based materials could be designed. Furthermore, in drug discovery, the precise structural information can be used to design more effective drugs with targeted interactions. This is an example of how understanding protein structures can translate into innovative solutions across different domains.
Conceptual Framework for Future Expansion
A future expansion strategy should be adaptable and scalable. A modular design, allowing for easy addition of new data types and methodologies, is crucial. This will ensure the database remains relevant and useful as new biological information emerges. The framework should prioritize data quality control and validation to maintain the accuracy and reliability of the database. This robust framework will facilitate a dynamic and evolving database, constantly adapting to new knowledge and methodologies.
Furthermore, an open-access policy is vital for fostering collaboration and accelerating research in various fields.
Technical Aspects and Implementation
The AlphaFold Protein Database’s success hinges on its robust technical infrastructure. This section delves into the intricate details behind its maintenance, data processing, storage, and retrieval, demonstrating the sophisticated engineering behind this powerful resource. The scalability and visualization capabilities are key elements ensuring continued utility for the scientific community.
Technical Infrastructure
The database’s infrastructure is designed with high availability and performance in mind. Distributed computing systems, leveraging cloud-based resources, are employed to handle the massive dataset. This allows for parallel processing of data, significantly reducing the time needed for tasks like model building and structure prediction. Redundancy and failover mechanisms are implemented to guarantee continuous access to the database.
Data Processing Algorithms and Tools
The core of AlphaFold’s success lies in its sophisticated algorithms. These algorithms use deep learning models, trained on massive datasets of protein sequences and structures, to predict the 3D structures of proteins. These models are continuously refined and improved, leading to increasingly accurate predictions. Specialized tools are developed and used to ensure data quality, handling issues like missing data, noise, and outliers.
These tools include filtering and validation procedures that ensure the accuracy of the predicted structures.
Data Storage and Retrieval Mechanisms
The database employs optimized storage mechanisms for efficient data access. Data is stored in a structured format, enabling quick retrieval and querying. Advanced indexing techniques are utilized to allow for rapid searches based on protein sequences, structures, or other relevant attributes. Data compression algorithms are employed to reduce storage space requirements while maintaining data integrity. The use of optimized query languages and interfaces makes it easy to retrieve the necessary information.
Scalability for Increasing Data Volumes
The database’s design ensures scalability to accommodate the ever-growing volume of protein data. The infrastructure is modular, allowing for easy expansion and adaptation to future needs. The use of distributed computing and cloud-based resources allows for effortless scaling to accommodate larger datasets. The system architecture allows for seamless integration of new data without disrupting existing functionality. This scalability is crucial for the ongoing development and maintenance of the database.
Visualization Tools for Protein Structures
The database provides various visualization tools to display protein structures. These tools are designed for easy exploration and understanding of the intricate details of protein structures. Interactive 3D models allow users to rotate, zoom, and manipulate the structures to study different aspects. The ability to overlay multiple structures or to highlight specific regions is a key feature for comparative studies.
Tools enabling the display of secondary structures (alpha-helices, beta-sheets) and important amino acid residues, along with the ability to identify and label specific domains, enhance the usability of the database. A table summarizing these visualization capabilities follows:
| Visualization Feature | Description |
|---|---|
| Interactive 3D Models | Allow users to rotate, zoom, and manipulate protein structures for detailed analysis. |
| Overlaying Multiple Structures | Facilitates comparative studies by displaying multiple protein structures simultaneously. |
| Highlighting Specific Regions | Allows users to focus on specific parts of the protein structure for detailed analysis. |
| Secondary Structure Display | Highlights alpha-helices and beta-sheets for understanding protein folding patterns. |
| Amino Acid Residue Identification | Enables users to identify and label key amino acid residues involved in specific functions. |
| Domain Visualization | Allows users to identify and label protein domains for studying their functional roles. |
Final Thoughts
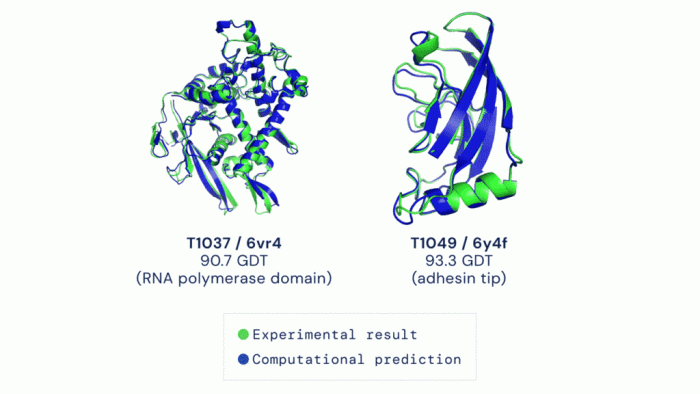
In conclusion, the DeepMind AlphaFold protein database alphabet is a transformative resource for biological research. Its ability to predict protein structures with remarkable accuracy is opening new avenues for understanding life’s complexities and driving progress in various scientific fields. While challenges remain, the database’s potential for future development and integration with other resources is immense. This resource is poised to shape the future of biology and beyond.